MoMI-G tools¶
The MoMI-G package includes a set of scripts (MoMI-G tools) that converts a VCF file into the variation graph format.
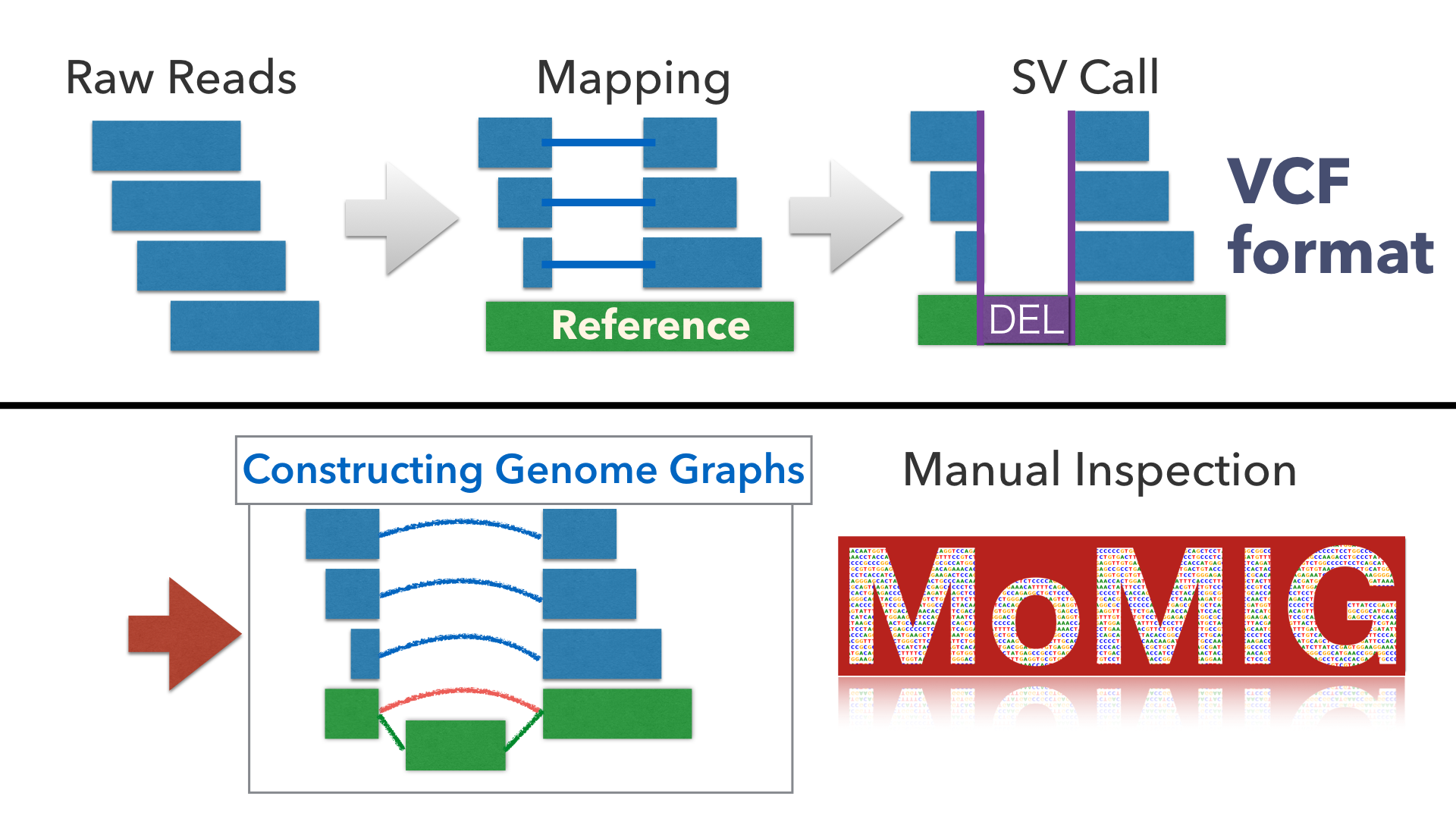
If you want to run MoMI-G with your own dataset, use our custom scripts scripts/vcf2xg.sh to generate pcf and xg dataset. It requires VG, ruby, bash, and samtools.
| Software | Dataset | Supported SV type |
|---|---|---|
| Sniffles | for PacBio/Nanopore | INV/DEL/DUP/TRA/(INS) |
| 10X LongRanger | for 10X | INV/DEL/DUP/TRA |
| SURVIVOR | for merging SV calls | INV/DEL/DUP/TRA |
$ bash vcf2xg.sh test.vcf test_output /bin/vg hg[19|38]
Otherwise, you can specify your own reference file as follows.
$ bash vcf2xg.sh test.vcf test_output /bin/vg /data/hs37d5.fa
You can use singularity instead.
$ singularity -s run docker://momigteam/momig-tools:master test.vcf test_output vg hg38
After that, these files are required to be mounted on static/ folder. Also, you should modify config.yaml and Dockerfile.backend in MoMI-G directory.
- static/
- config.yaml: a configuration file
- *.xg: an index of a variation graph, generated by [vg](https://github.com/vgteam/vg)
- *.pcf: a pair of coordinate format file: required to display variants on Feature Table or Circos Plot.
- *.gam(optional): read alignments on the graph
- *.gam.index(optional): index of gam
This is an example of config.yaml and Dockerfile. \* is just an example and you need to replace it to the actual file name.
bin:
vg: "vg"
vg_tmp: "vg"
vg_volume_prefix: ""
graphviz: "dot"
fa22bit: "faToTwoBit"
bigbed: "bedToBigBed"
reference:
chroms: "static/GRCh.json"
data:
- name: "hg19"
features:
- name: 'gene_annotation'
url: "static/gencode.v27lift37.basic.annotation.gff3"
chr_prefix: "chr"
- name: "hg38"
features:
- name: 'gene_annotation'
url: "static/gencode.v27.basic.annotation.gff3"
chr_prefix: "chr"
data:
- name: "data"
desc: "2019/12/19"
chr_prefix: "chr"
ref_id: "hg38"
source:
xg: "static/\*.xg" # To be rewritten
csv: "static/\*.pcf" # To be rewritten
# gam: "static/b.gam"
# gamindex: "static/b.gam.index"
features: []
static_files: []
# Specify the version you used to build xg index.
FROM quay.io/vgteam/vg:v1.14.0 as build
# frontend container
FROM momigteam/momig-backend
COPY --from=build /vg/bin/vg /vg/bin/
# Move these files into /vg/static/ folder.
COPY static/\*.xg /vg/static/
COPY static/\*.pcf /vg/static/
COPY static/config.yaml /vg/static/
EXPOSE 8081
CMD ["./graph-genome-browser-backend", "--config=static/config.yaml", "--interval=1500000", "--http=0.0.0.0:8081", "--api=/api/v2/"]
If you use the later VG (version >= 10), gam.index file is no longer used. Please use sorted.gam.gai instead (bam2gam.sh generates gam.gai file).
- static/
- config.yaml: a configuration file
- *.xg: an index of a variation graph, generated by [vg](https://github.com/vgteam/vg)
- *.pcf: a pair of coordinate format file: required to display variants on Feature Table or Circos Plot.
- *.sorted.gam(optional): read alignments on the graph
- *.sorted.gam.gai(optional): index of gam
Then, run the MoMI-G backend.
$ docker build -t momig-custom-backend -f Dockerfile.backend .
$ docker run --init -p 8081:8081 -v `pwd`/static:/vg/static momig-custom-backend
At last, run the MoMI-G frontend.
$ sed -e "s/\"target/\"target_/g" -e "s/\_target/target/g" -i.bak package.json
$ yarn
$ yarn start